1 Pediatra. UGC Delicias. Málaga. España.
2 Pediatra. CAP Sant Ildefons. Cornellà de Llobregat. Barcelona. . España.
Los estudios experimentales se utilizan para evaluar la eficacia y la efectividad de intervenciones terapéuticas, preventivas o educativas. Hay diferentes tipos de estudios experimentales, pero, actualmente, el ECA se reconoce como el tipo de estudio que proporciona el nivel más alto de evidencia. Cuando no se puede realizar este tipo de investigación, existen estudios cuasiexperimentales, con menor grado de validez, donde puede no haber aleatorización o un grupo de control1,2.
Al tratarse de investigación en humanos y aún más cuando se realiza en niños, es muy importante el diseño de los estudios, de acuerdo con los estándares científicos aceptados tanto éticos como metodológicos3, suponiendo todo ello un mayor desafío que la investigación con adultos4.
La legislación sobre investigación con población pediátrica se ha desarrollado principalmente en las dos últimas décadas tanto en EE. UU. como en Europa. La Organización Mundial de la Salud (OMS) ha reconocido los riesgos que supone el uso de fármacos en población pediátrica que previamente no se han ensayado en ella, por lo que desde el Parlamento Europeo se estableció la necesidad de disponer de mejores medicinas en Pediatría, con el desafío específico de desarrollar medicamentos para enfermedades que solo afectan a los niños o para enfermedades que se manifiestan de manera diferente en adultos y niños, estableciendo la normativa a desarrollar en todos los países de la Unión, garantizando la autorización de medicamentos específicamente desarrollados para ser utilizados en la población pediátrica, incentivando la investigación en estas edades mediante estudios multicéntricos5,6.
El conocimiento de la correcta elaboración de un estudio experimental trasciende el mero interés de la investigación clínica para contribuir a desarrollar un sentido crítico en los lectores, permitiendo una mejor valoración de los resultados publicados7-10.
Para aumentar la transparencia en la investigación clínica, el registro de libre acceso para los ECA a su inicio (http://clinicaltrials.com) y la publicación de sus resultados, debería ser obligatorio a escala mundial y contribuiría a evitar el sesgo de publicación, debido a la publicación de resultados favorables y supresión de resultados negativos11,12.
La declaración CONSORT 10 (Consolidated Standars of Reporting Trials, www.equator-network.org/reporting-guidelines/consort/13,14 ha supuesto un gran avance para que la publicación de los ECA sea más rigurosa y recoja de forma adecuada los aspectos metodológicos.
Los ensayos clínicos aleatorizados con un diseño y un desarrollo apropiados constituyen el “patrón oro” en la evaluación de las intervenciones en los cuidados sanitarios13.
Resulta de vital importancia establecer módulos y programas de investigación durante la licenciatura, en la formación especializada y durante la carrera profesional12.
El diseño del ensayo clínico comienza con la formulación de la pregunta de investigación y la identificación de posibles fuentes de sesgo.
La pregunta de investigación debe articular explícitamente la población, la intervención, la comparación y los resultados (PICO) que se medirán. Este aspecto es tratado de forma específica en otro de los artículos del monográfico.
Un ensayo clínico permite una evaluación experimental sobre la eficacia y la efectividad de intervenciones terapéuticas, preventivas o educativas. En el caso concreto de los medicamentos, técnicas diagnósticas o terapéuticas aplicadas a humanos, existe una regulación específica recogida en el RD 1090/2015. Según este RD, se definiría un ensayo clínico con medicamentos como “toda investigación relativa a personas destinada a:
Los ensayos clínicos deberán realizarse de acuerdo con la Declaración de Helsinki sobre los principios éticos para las investigaciones médicas en seres humanos, aprobada por la Asamblea General de la Asociación Médica Mundial y teniendo en cuenta el Convenio de Oviedo para la protección de los derechos humanos y la dignidad del ser humano con respecto a las aplicaciones de la Biología y la Medicina, así́ como a cualesquiera otras normas que pudieran resultar de aplicación15.
Los ensayos clínicos se deben realizar en condiciones de respeto a los derechos fundamentales de la persona y a los postulados éticos que afectan a la investigación biomédica16. En España, se establece que ningún ensayo clínico podrá realizarse sin el informe previo favorable de un comité de ética de la investigación (CEI), el RD 1090/2015 define dentro de CEI el subgrupo de comités de ética de la investigación con medicamentos (CEIm), otorgando a estos últimos la responsabilidad adicional de evaluar los estudios clínicos con medicamentos o con productos sanitarios, que será independiente de los promotores, investigadores y de las autoridades sanitarias, regulado a nivel Europeo por el Reglamento (UE) n.º 536/2014 del Parlamento y Consejo Europeo.
El Real Decreto 1090/2015 por el que se regulan los ensayos clínicos con medicamentos en España dedica algunos artículos a la investigación en Pediatría. Determina que es necesario fomentar la investigación clínica de medicamentos huérfanos y de medicamentos destinados al tratamiento de grupos de población como niños, mujeres y ancianos que tradicionalmente han estado poco representados en la investigación clínica. Es importante valorar la posible aparición de efectos adversos o peligrosos y en este sentido cabe destacar la importancia de las medidas que protegen a los niños de experimentaciones poco éticas, sin menoscabo de las actuaciones que permitan avances en la terapéutica pediátrica17.
La realización de ensayos clínicos en Atención Primaria, debido a su elevado coste, al tiempo que requieren y a las barreras éticas, deben contemplarse bajo el auspicio de un organismo que permita una participación multicéntrica.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) incluirá en la base de datos europea de ensayos clínicos (EudraCT) los datos relativos a los ensayos clínicos con medicamentos de uso humano que se lleven a cabo en el territorio nacional y mantendrá el sistema de información de dichos EC publicándolos en su página web18.
El registro Español de Ensayos clínicos (REec) contiene la información de todos los ensayos clínicos con medicamentos autorizados en España desde el 1 de enero de 2013 y próximamente incluirá, además, información sobre estudios observacionales con medicamentos de forma obligatoria y otros tipos de investigaciones clínicas de forma voluntaria (https://reec.aemps.es/reec/public/web.html)
Este diseño es el más riguroso para la evaluación de cualquier intervención; para llamarlo ECA debe cumplir con cuatro características principales:
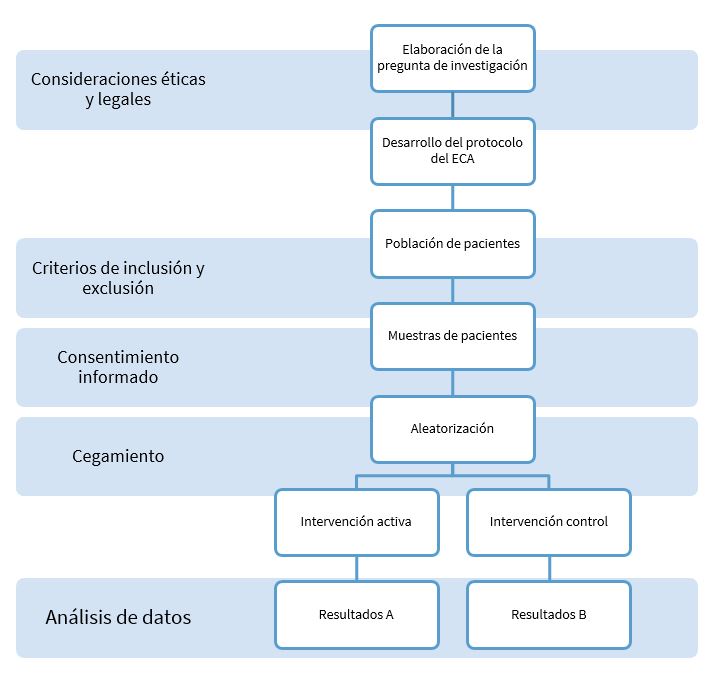
El valor de este diseño en el campo de la biomedicina se aprecia, por ejemplo, al observar que la mayoría de las recomendaciones terapéuticas y guías de práctica clínica se basan en las pruebas proporcionadas por ECA, además las autoridades sanitarias obligan a la realización de un ECA para demostrar la eficacia y seguridad de un nuevo fármaco antes de su comercialización. Este tipo de diseño nos acerca, de forma más directa, a la noción de causalidad entre una intervención y unos resultados concretos20 (Figura 1).
Figura 1. Diseño de los ECA20. Mostrar/ocultar
El sujeto o participante de un ECA es la persona sana o enferma que participa en él, después de haber otorgado libremente su consentimiento informado.
La definición del objetivo del ensayo hace referencia a la población diana a la que se desea poder extrapolar los resultados. Sin embargo, el estudio se lleva a cabo sobre una población definida por unos criterios de selección especificados a priori (población experimental), de la que se obtendrán los sujetos que finalmente participarán en el ensayo.
La utilización de criterios de inclusión y exclusión estrictos conduce a la obtención de una muestra homogénea, lo que aumenta la validez interna del estudio, pero que, al alejar la población de estudio de la diana, limita su capacidad de generalización o extrapolación.
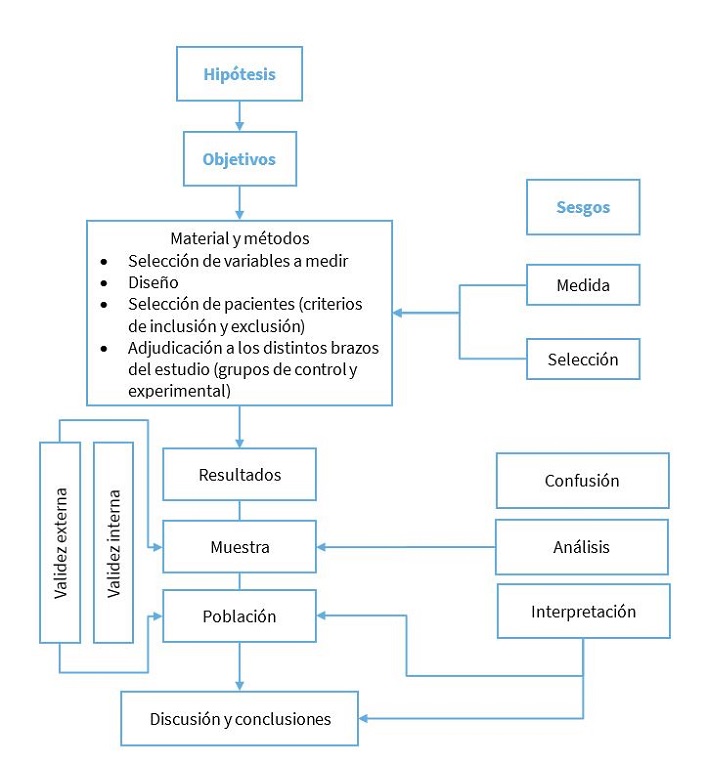
Por otro lado, si se definen criterios muy amplios, la población de estudio será más representativa de la diana y las posibilidades de generalizar los resultados serán mayores, pero, al ser más heterogénea, será más difícil detectar una respuesta al tratamiento y se requerirá un mayor número de individuos (Figura 2).
Figura 2. Esquema del desarrollo de un ensayo clínico. Validez. Sesgos. Mostrar/ocultar
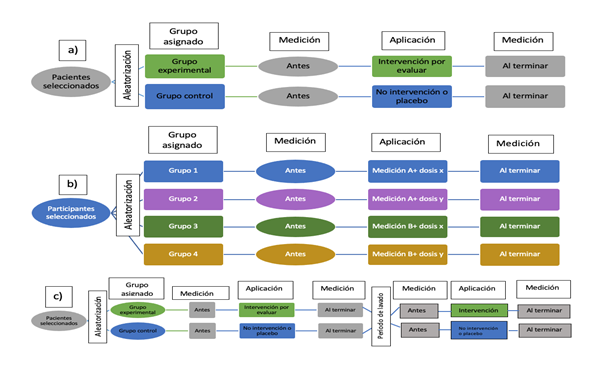
Tanto la selección de sujetos como los periodos de tratamiento y seguimiento han de tener lugar simultáneamente en todos los grupos. En la gran mayoría de los casos es la única forma científicamente válida para evaluar la eficacia y seguridad de una intervención terapéutica. Estos ensayos pueden ser:
Figura 3. Tipos de ensayos clínicos controlados y aleatorizados (ECCA). A) ECCA clásico. B) ECCA factorial. C) ECCA cruzado. Mostrar/ocultar
La determinación de un tamaño muestral adecuado pretende dotar a la muestra del poder estadístico suficiente para que si existen diferencias no debidas al azar entre los dos grupos puedan detectarse. Para ello, además de tener en cuenta la homogeneidad de las poblaciones participantes, es necesario valorar la cuantía de las diferencias que se pretende poner de manifiesto, los errores de tipo I y II que se consideran aceptables y la proporción esperada de pacientes que abandonarán el estudio. Así pues, la decisión sobre el tamaño de la muestra es muy importante. Una muestra demasiado grande implica un gasto excesivo de recursos, y, por el contrario, una muestra demasiado pequeña disminuye la utilidad de los resultados.
Es imprescindible calcular el tamaño necesario de la muestra según los objetivos del estudio, el diseño planteado y el tipo de variables, así como decidir qué técnica de muestreo se utilizarán para seleccionar a los sujetos16.
El muestreo es el proceso que permite extraer una muestra de una población y permitirá realizar inferencias sobre una población, estudiando una muestra extraída de ella.
Los errores o sesgos en la recogida de la muestra pueden desautorizar los resultados al no ser totalmente representativa de la población a estudiar.
Tras la elección de una muestra representativa, se procede a realizar la propuesta de participación a los sujetos integrantes, así como a la presentación del consentimiento informado (CI), requisito fundamental para poder ser incluido en el ensayo. Todo estudio de investigación en el ámbito clínico debe, según la normativa, incluir un documento de CI, el cual consta de una Hoja de Información al Paciente (HIP) y del documento donde este deberá aceptar con su firma la participación en el estudio2,22,25. En el artículo 4 del RD 1090, respecto al consentimiento informado de los participantes, especifica que, en el caso de menores o personas con la capacidad modificada para dar su consentimiento, habiendo dado el consentimiento su representante legalmente designado, cuando estos alcancen o recuperen la capacidad de consentir, deberá recabarse su consentimiento para continuar participando en el ensayo clínico.
El Young Person's Advisory Group del Hospital Sant Joan de Déu de Barcelona llevó a cabo un proyecto de análisis del formato y contenido de los documentos de consentimiento informado que se utilizan en la práctica. Conscientes de que en muchas ocasiones este documento no se adapta a las necesidades de información y comprensión de los niños, este grupo ha elaborado un informe de recomendaciones para el diseño del documento de asentimiento para pacientes pediátricos participantes en un ensayo clínico (Tabla 1)19.
Tabla 1. Recomendaciones para el diseño del consentimiento informado/asentimiento de ensayos clínicos con pacientes pediátricos del grupo Young Person's Advisory Group del Hospital Sant Joan de Déu (Barcelona). Mostrar/ocultar
La fortaleza principal de un ensayo clínico es la adecuada aleatorización. Consiste en la asignación de los sujetos participantes a los diferentes grupos de intervención por un mecanismo únicamente debido al azar, de modo que ni el investigador ni el sujeto pueden influir y no conocen el grupo al que son asignados.
Permite una distribución equilibrada de las características de los pacientes a los grupos de tratamiento, generando grupos comparables respecto a cualquier condición conocida o no –variables de confusión– que pudiera afectar al resultado, así la diferencia principal entre los grupos será la intervención recibida. Permite crear grupos similares al inicio del ensayo, en cuanto al riesgo de presentar el evento que se espera modificar con la intervención.
La eficacia de la aleatorización depende del tamaño muestral del estudio, así en muestras pequeñas, aunque sean adecuadamente aleatorizadas, existen más posibilidades de que la distribución de las características de los grupos de intervención no sea totalmente homogénea.
La asignación aleatoria, cuando se realiza con un tamaño muestral adecuado, equilibra los grupos respecto de los factores pronósticos que puedan determinar un mayor o menor riesgo de presentar el resultado, ya sean conocidos o no26.
Existen diferentes métodos de aleatorización, aunque los más frecuentemente empleados son la aleatorización simple, aleatorización por bloques y aleatorización estratificada.
Un ejemplo de aleatorización simple sería lanzar una moneda al aire, o de forma más sofisticada, el empleo de tablas de números aleatorios, o la generación de números aleatorios mediante un programa informático. Son sistemas sencillos, pero en muestras pequeñas no garantizan el equilibrio en el tamaño de los grupos, ni la distribución homogénea de los factores de confusión. Estos problemas se resuelven utilizando la aleatorización por bloques, en la que se definen bloques de sujetos de un tamaño predeterminado en los que, por diseño, se fuerza a que el número de sujetos asignado a cada una de las intervenciones en estudio sea igual. Una vez constituidos los bloques se aleatorizan uno a uno los sujetos de cada bloque.
En la aleatorización estratificada se definen previamente los factores pronósticos por los que se desea estratificar (por ejemplo, sexo: hombre/mujer, y estado civil: soltero/casado/separado/viudo). Se constituyen tantos estratos como el producto del número de categorías de cada uno de los factores considerados. Cada vez que se incorpora un nuevo sujeto al estudio se le asigna directamente a su estrato correspondiente (mujer soltera), y dentro de cada estrato se realiza un proceso de aleatorización independiente.
Además de la posibilidad de homogeneizar los grupos, evitando que sean otras variables diferentes de la intervención las que condicionen los resultados obtenidos, la aleatorización permite utilizar técnicas de enmascaramiento para evitar el denominado sesgo de selección. Este sesgo ocurre cuando, o bien el investigador encargado de realizar la asignación, o bien el sujeto, tienen alguna influencia sobre el grupo al que será incorporado. Es fácil que el investigador conozca qué tratamiento corresponde a cada uno de los grupos, como es el caso de intervenciones que asocian efectos adversos característicos. El interés personal del investigador en el éxito del nuevo tratamiento puede influir en que trate de evitar que los sujetos con mejor pronóstico o mayores expectativas de mejoría vayan al grupo control.
Se definen como aquellos procedimientos realizados con el fin de que algunos de los sujetos relacionados con el estudio (equipo investigador, participantes, etc.) no conozcan algunos hechos u observaciones (básicamente el tratamiento que recibe cada sujeto) que pudieran ejercer un cambio en sus acciones o decisiones y sesgar los resultados. Un estudio que no utiliza técnicas de enmascaramiento se denomina ensayo abierto.
El cegamiento permite que cualquier grado de imparcialidad sobre alguna de las intervenciones en estudio por parte del investigador, o bien del sujeto participante (autosugestión, confianza en una nueva intervención, etc.), pueda influir sobre los resultados o la interpretación de estos. De este modo, se evita el denominado sesgo de realización, que se da cuando existen diferencias en el trato o la atención sanitaria que reciben los sujetos en función del grupo al que pertenezcan. Si el grupo asignado es desconocido, todos los sujetos recibirían exactamente la misma atención, excepto la propia intervención.
Se ha determinado que los ECA que no especifican un adecuado ocultamiento pueden sobreestimar la medida del efecto en un rango del 30 al 40%, en relación con aquellos que sí explicitan métodos con esta finalidad.
La técnica del simple ciego consiste en que los investigadores, o más frecuentemente los propios participantes, desconozcan qué intervención recibe cada individuo.
Si los investigadores conocen quien recibe cada intervención, o los participantes saben qué tratamiento reciben, existe la posibilidad de que se examine con mayor minuciosidad cualquier respuesta, o se pregunte con más detalle por los posibles efectos secundarios de alguno de los tratamientos. Estas preferencias se evitan con la técnica del doble ciego, donde tanto los pacientes como los investigadores desconocen el tratamiento administrado.
La técnica del triple ciego, en la que, además, hay otras personas que también desconocen el tratamiento que recibe cada sujeto, ya sea el profesional estadístico que analizará los resultados, o la persona responsable de decidir si se suspende un tratamiento por la aparición de reacciones adversas o si debe interrumpirse prematuramente el ensayo (Tabla 2).
Tabla 2. Ejemplos de tipos de enmascaramiento utilizados en ensayos clínicos10. Mostrar/ocultar
Aunque lo más deseable para la realización de un ECA es seguir una metodología, de doble ciego, esta no siempre es posible. En el caso de intervenciones farmacológicas, los efectos secundarios o el perfil de toxicidad de algunos fármacos impide su cegamiento, así́ como en aquellos casos en los que no es posible disponer de una formulación galénica adecuada que actúe como grupo control placebo. Tampoco es posible cuando suponga riesgos innecesarios para el paciente, como en los casos en los que tendría que ser sometido a intervenciones quirúrgicas simuladas, o a administraciones repetidas de placebo por vía parenteral. Por último, en todos aquellos casos en los que por cualquier circunstancia el diseño doble ciego puede perjudicar la relación entre el médico y el paciente, no es conveniente llevarlo a cabo.
Cuando estos métodos no pueden llevarse a cabo, puede utilizarse la técnica de evaluación enmascarada de la respuesta, o del evaluador ciego. Consiste en que la persona que ha de medir la variable de respuesta desconozca el grupo al que pertenece cada uno de los sujetos, con la finalidad de que la medición se realice e interprete de la misma forma para cada grupo.
Para establecer la validez del ciego en un ECA, más importante que la descripción acerca de si se trata de doble o triple ciego, es el detalle explícito de todos los que lo fueron en el desarrollo del ensayo, los métodos utilizados para lograrlo y su éxito o fracaso26.
Otro aspecto metodológico importante en la realización de un ECA es efectuar una correcta descripción de las pérdidas y abandonos experimentados durante su transcurso, es decir, recoger el número de sujetos que abandonan el estudio antes de su finalización, el grupo al que pertenecen y, si es posible, la causa del abandono o pérdida (aparición de evento adverso, falta de eficacia del tratamiento, etc.). Cada una de las causas tendrá un significado diferente en la valoración de la eficacia de la intervención en estudio y, por tanto, disponer de esta información es muy importante para que no se produzca el denominado sesgo de seguimiento.
A partir de esta información hay diferentes estrategias para analizar los datos y llegar a resultados válidos. Una de las opciones recomendadas es la realización de un análisis por intención de tratar. Según esto, cada paciente es analizado en el grupo al que fue asignado al inicio del estudio, independientemente de que no terminara la intervención que correspondía a ese grupo. Este tipo de análisis, además de permitir controlar el sesgo de seguimiento, es la única estrategia que conserva las ventajas adquiridas mediante la asignación aleatoria de los sujetos, creando dos grupos comparables en todas las variables excepto en la intervención recibida, y es la estrategia que más se aproxima a la realidad de la práctica clínica diaria, donde los pacientes con frecuencia incumplen o rechazan los tratamientos prescritos.
Hay estudios que muestran resultados beneficiosos de una intervención al analizar solo los pacientes que completan o se adhieren al protocolo (análisis por protocolo o por tratamiento); sin embargo, al analizar por intención de tratar, los beneficios no son tan evidentes.
Los estudios realizados en niños precisan consideraciones especiales. Los investigadores deben medir los resultados obtenidos de la forma más rigurosa posible. Además, hay que considerar que los niños y los adultos responden de forma particular a los tratamientos realizados. Se debe contemplar la realización de grupos de edad para una mejor valoración de los ensayos clínicos en Pediatría27.
Para salvaguardar la garantía de la correcta elaboración de los estudios de investigación, y en un intento de mejorar la calidad de su publicación, han surgido guías de ayuda, siendo al menos 25 escalas y nueve listas o checklists las que tratan de evaluar la validez y la "calidad" de los ensayos clínicos. En el caso de estudios experimentales, la declaración CONSORT (Consolidated Standars for Reporting of Trials) resulta recomendable para mejorar la calidad de la escritura y la publicación de los ensayos clínicos26. La declaración o guía CONSORT inicial, permitió un aumento en la calidad de la comunicación de los ECA, y ha sido revisada en varias ocasiones28, siendo la más reciente la publicada en el año 201029. Incorpora en la actualidad una lista de comprobación o cuestionario de 25 ítems que se consideran críticos y que por tanto deberían incluirse en todo informe de un ensayo clínico junto con el diagrama de flujo, en el que se establece los cuatro apartados de un ECA como son el reclutamiento, la intervención, el seguimiento y el análisis. En dicho diagrama debe constar la identificación sobre el seguimiento de los pacientes incluidos en el estudio, necesario para considerar la validez interna y la aplicabilidad de los resultados obtenidos, que muestra el flujo de individuos participantes a lo largo del estudio30.
Estas herramientas se centran en el ECA de dos grupos paralelos. Otros diseños, como los ensayos con aleatorización de conglomerados (en los que se aleatorizan grupos o "conglomerados" de individuos), o los ensayos de no inferioridad, o los ECA sobre tratamientos no farmacológicos, pueden requerir información adicional y existen extensiones de CONSORT específicas para ellos que pueden consultarse en la página web del grupo CONSORT (http://www.consort-statement.org).
El grupo CONSORT integra una red internacional, la Red EQUATOR Network (Enhancing the Quality and Transparency Of health research), que promueve y facilita la publicación de estudios de investigación de salud. En el sitio Web de la Red (www.equator-network.org), se aportan directrices sobre escritura científica o ética de la investigación31,32.
Se puede consultar un catálogo de las diferentes extensiones de la declaración CONSORT, así como más de 90 listas de comprobación desarrolladas para cada tipo de estudio33,34.
Ledesma Albarrán JM, Balaguer Martínez JV. Estudios experimentales. Ensayo clínico aleatorizado. FAPap Monogr. 2021;6:45-54