1 Pediatra. CS de Pasajes San Pedro. Pasajes. Gipuzkoa. España.
En el presente artículo, tras realizar un repaso de los aspectos históricos relacionados con la regulación ético-legal de la investigación biomédica, se analizan las normativas, tanto internacionales como europeas y nacionales, sobre la regulación de la experimentación con seres humanos.
A continuación, se expone el proceso de evaluación de un proyecto de investigación, en el que se tienen en cuenta el diseño metodológico, la idoneidad de los investigadores y la adecuación de las instalaciones, el balance riesgo/beneficio del estudio y los métodos que vayan a utilizarse para informar a los sujetos que participan en la investigación, con el fin de obtener su consentimiento informado. La investigación en menores de edad es un caso especial, al precisar un consentimiento de sus tutores y un asentimiento del niño, que varía según la edad de este.
Finalmente, se explican los diferentes documentos que hay que presentar a un Comité de Ética de la Investigación para solicitar la aprobación de un proyecto de investigación o un ensayo clínico.
La participación de seres humanos en investigación biomédica ha planteado desde tiempos remotos serios dilemas morales. Inicialmente, se suponía que los propios médicos realizaban una autorregulación de sus actividades clínicas y de investigación. Parecía que el juramento Hipocrático garantizaba dicha autorregulación.
“Estableceré el régimen de los enfermos de la manera que les sea más provechosa según mis facultades y entender…”, dice. Por lo tanto, era el propio médico el que se consideraba como único capacitado para juzgar acerca de la idoneidad de su actividad que tenía por objetivo la búsqueda de la salud.
El conocimiento de las atrocidades perpetradas en los campos de concentración nazis motivó el desarrollo de los llamados códigos históricos, el de Nuremberg y el de Helsinki, documentos que han facilitado que los profesionales de la biomedicina puedan ejercer su actividad con respeto a una serie de principios éticos y jurídicos. A continuación, veremos las características de ambos textos y el momento en el que fueron redactados.
El Código de Nuremberg (1946) recoge una serie de principios que rigen la experimentación con seres humanos. Fue el resultado de las deliberaciones de los responsables médicos, al final de la Segunda Guerra Mundial, tras el tratamiento inhumano que habían dado los médicos nazis a los prisioneros de los campos de concentración.
Establece en su artículo primero que para la aplicación de cualquier medida que tenga carácter experimental es absolutamente esencial el consentimiento voluntario. Y continúa: “el deber y la responsabilidad de asegurar las condiciones de calidad del consentimiento recaen sobre cada médico que inicia, dirige o participa en el experimento”. Este fue el paso fundamental.
La Declaración de Helsinki de la Asociación Médica Mundial se adoptó en la 18.ª Asamblea, en 1964. Supuso el traslado de los principios del Código de Nuremberg al ámbito de los principios éticos de las organizaciones médicas. La Declaración de Helsinki ha promulgado los principios éticos que sirven para orientar a los médicos y a otras personas que realizan investigación médica en seres humanos. Estos principios establecen que el protocolo de la investigación debe enviarse, antes de comenzar el estudio, a un Comité de Ética de Investigación, que será independiente del investigador, del patrocinador o de cualquier otro tipo de influencia indebida, y que deberá considerar las leyes y reglamentos del país donde se realiza la investigación, y las normas internacionales vigentes.
La declaración ha tenido cinco modificaciones importantes que han ido limitando y concretando más los principios enunciados. Así, por ejemplo, en la 29.ª Asamblea de Tokio se introdujo por primera vez que deberían ser comités independientes de los protocolos de investigación los que valoraran el mismo. En Venecia (1983) se introdujo una referencia del consentimiento a menores de edad. En 1989, en Hong Kong, se precisaron las condiciones de independencia de los comités y su conformidad con las leyes nacionales. La de 2000 supuso un cambio competo de las condiciones formales y estructurales de la Declaración cuyos pasos han dejado de llamarse “recomendaciones” para pasar a “principios” y, así, hemos llegado a la última revisión del año 2014 en Fortaleza (Brasil). En esta última revisión se han añadido principios que inciden en una mayor protección de los participantes en la investigación; así, por primera vez, la declaración exige la compensación y tratamiento de los daños relacionados con la investigación. Otra incorporación es la exigencia de enviar el informe final de los estudios, lo que facilita el seguimiento de las condiciones de realización de la investigación a los Comités de Ética de la Investigación1,2.
Y, desde 2016, la Declaración de Helsinki está complementada con la Declaración de Taipei sobre las consideraciones éticas sobre las bases de datos de salud y los biobancos3, que pretende cubrir la recopilación, el almacenamiento y el uso de la información y el material biológico identificables más allá de la atención individual de pacientes.
En la Tabla 1 se resumen las aportaciones de cada uno de los pasos dados en el control de la investigación en humanos.
Tabla 1. Evolución histórica de la regulación de la investigación médica. Mostrar/ocultar
Pero ni el Código de Nuremberg ni la Declaración de Helsinki tuvieron repercusión inmediata en la investigación con seres humanos. Es más, durante años posteriores se dieron casos de investigaciones que no tenían en cuenta para nada los principios éticos. Dos ejemplos muy claros son los estudios sobre la hepatitis en niños en la escuela Willowbrook y los estudios sobre la sífilis en Tuskegee.
Entre 1963 y 1966 en la escuela Willowbrook del estado de Nueva York para niños con trastornos mentales, 700 niños discapacitados fueron infectados intencionalmente, de forma oral o por inyección, con el virus de la hepatitis, para luego monitorizar los efectos de la ganmagobulina para combatir dicha enfermedad. Una protesta pública forzó el fin del estudio.
Entre 1932 y 1972, 400 pacientes de sífilis de raza negra y 200 hombres sanos fueron excluidos del tratamiento a pesar de que en 1947 se demostró que la penicilina era eficaz para el tratamiento de dicha enfermedad. Como resultado de dicho estudio, 40 pacientes fallecieron de sífilis, 100 fallecieron de complicaciones médicas relacionadas con la sífilis, 40 mujeres resultaron infectadas y 19 niños padecieron sífilis congénita. Inicialmente, el estudio iba a durar 6-8 meses. En 1928 se publicó un estudio retrospectivo en Oslo, en sífilis no tratada y en Tuskegee decidieron realizar un estudio similar pero prospectivo. A pesar de que en 1947 se descubrió la eficacia de la penicilina, decidieron continuar con el estudio ocultando esa información a los pacientes. Además, se realizaron otras acciones fuera de toda ética; por ejemplo, se realizaron punciones lumbares sin función terapéutica que los doctores presentaban en una carta como “la última oportunidad de un tratamiento especial y gratuito”. Igualmente, se les solicitaba permiso para realizar la autopsia diciendo que era necesario aceptar para recibir el seguro que cubría los gastos del sepelio. El Dr. Peter Buxtum, que descubrió este caso, acudió a la prensa. La noticia apareció en el Washington Star en 1972, Al día siguiente fue portada del New York Times. Inmediatamente el estudio se paró.
Más cerca de nosotros, hace pocos años se descubrió que, además de estos dos casos ya relatados, hubo otro estudio parecido sucedido en Guatemala en la década de los 40. La Investigadora Susan Reverby, que estaba realizando una investigación acerca de lo sucedido en Tuskegee, en los archivos de la Universidad de Pittsburgh encontró datos sobre otra investigación coordinada por el mismo médico y realizada en Guatemala. Este es el relato de la prensa: “Entre 1946 y 1948, un grupo de médicos estadounidenses, dirigidos por John Carles Cutler, bajo el patrocinio directo de la secretaría de salud del gobierno estadounidense, inoculó sífilis y gonorrea, sin darles ninguna información, a soldados, prostitutas, prisioneros y hasta a niñas de un hospicio. Fueron 696 los guatemaltecos infectados para probar con ellos los efectos curativos de la penicilina en relación con estas enfermedades venéreas”.
Pero sin ir tan lejos en el tiempo, recientemente, en año 2011 ha habido una alerta sobre el aumento de muertes en ensayos clínicos en la India. El gobierno de India ha admitido la muerte de 1725 personas en ensayos clínicos con medicamentos en los últimos cuatro años. El número de fallecidos ha sido de 132 en 2007, 288 en 2008, 637 en 2009 y 688 en 2010. En India no existe un sistema independiente de auditoría para investigar las causas de dichas muertes. El doctor Chandra Gulhati, que actualmente está investigando la causa de 81 muertes ocurridas en ensayos clínicos en la ciudad de Indore dice que “como los investigadores son contratados por las empresas farmacéuticas que van a realizar el ensayo, existen dudas sobre su objetividad”. La mayor parte de las investigaciones se han realizado por encargo de compañías internacionales que realizan sus ensayos en la India porque su coste es un 80% menor que en países desarrollados. Existen otras deficiencias, así, por ejemplo, a pesar de que la regulación exige la firma de un consentimiento informado en algunas ocasiones se ha comprobado su ausencia4.
Vemos por lo tanto que la vigilancia sobre el control de las investigaciones debe ser constante y que ante la exigencia de un mayor control en los países desarrollados las compañías farmacéuticas buscan realizar sus investigaciones en lugares donde el control es menos estricto, por lo que las normas deben ser universales. El ser humano debe estar protegido para su participación libre y sin riegos en los estudios de investigación en cualquier parte del mundo.
Tras esa historia, antigua y reciente, un poco oscura, en la que los principios y declaraciones sobre la protección de los sujetos en relación con la participación en las investigaciones médicas no se llevaba a la práctica, los estados fueron incorporando a sus legislaciones los principios citados, fundamentalmente, la Declaración de Helsinki. Lo ocurrido en los años 70 marcó notablemente la actitud de los profesionales de la salud ante la investigación, y el Departamento de Salud, Educación y Bienestar de los EE. UU., elaboró un informe en 1978 titulado "Principios éticos y orientaciones para la protección de los seres humanos en la investigación", que es conocido como Informe Belmont, porque fue en el Centro de Conferencias de dicha ciudad donde el documento fue elaborado, y establece tres principios básicos: el respeto a las personas, la beneficencia, es decir, el esfuerzo que tiene que realizar el investigador para asegurar el bienestar de las personas, y el de justicia, en el sentido de que no se prive a nadie de un tratamiento efectivo sin un motivo razonable (Figura 1).
Figura 1. Repercusión en la legislación de los principios éticos. Mostrar/ocultar
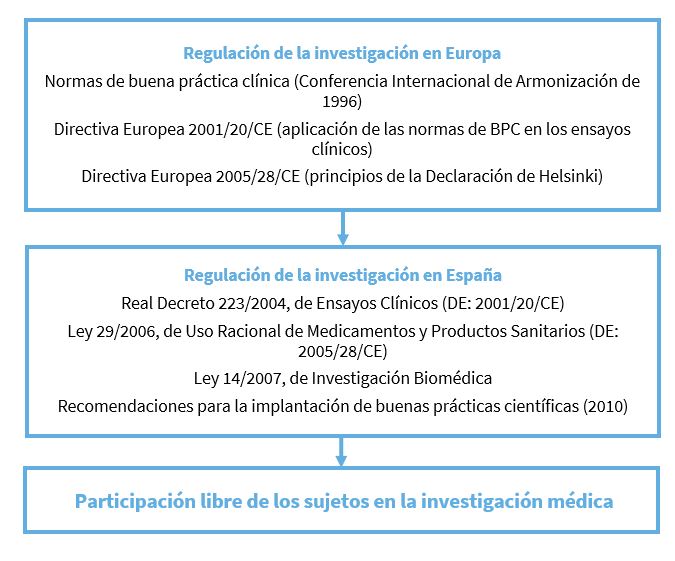
Los diferentes estados fueron estableciendo cada uno sus normas relativas al control de la investigación. En EE. UU., la FDA (Food and Drug Administration) publicó en 1977 y 1978 las “obligaciones del promotor y del investigador”; lo mismo sucedió a nivel europeo y de la OMS. Con el fin de tener unas normas con criterios unificados para todo el mundo surgió la Conferencia Internacional de Armonización (ICH), que, en su tercera edición, elaboró un documento oficial para unificar las normas de buenas prácticas clínicas (BPC) entre los países de la Unión Europea, Japón y EE. UU. Esta guía ICH de BPC fue aprobada en julio de 1996 por el Comité de Especialidades Farmacéuticas (CPMP) de la Unión Europea (CPMP/ICH/135/95)5, en 1997 es incorporado a la legislación japonesa y en el mismo año a la estadounidense. Como consecuencia de su incorporación a la legislación europea, a partir del 17 de enero de 1997, todos los ensayos clínicos que se realicen en la Unión Europea deben cumplir estas normas ICH.
Las Normas de Buena Práctica Clínica (BPC) establecen que los ensayos clínicos deben ser diseñados, realizados y comunicados de modo que se asegure que los datos son fiables y que se protegen los derechos y la integridad de los sujetos, manteniendo la confidencialidad de sus datos. Estas normas señalan las responsabilidades de los diferentes agentes implicados en cada una de las fases de planificación y ejecución de un ensayo clínico y requieren la existencia de unos procedimientos preestablecidos por escrito que se apliquen de forma sistemática en la organización, dirección, recogida de datos, documentación y verificación de los ensayos clínicos (procedimientos normalizados de trabajo).
El parlamento europeo aprobó el 4 de abril de 2001 la Directiva 2001/20/CE6 sobre la aplicación de las BPC en la realización de ensayos clínicos con medicamentos de uso humano con el objetivo de agilizar la tramitación de las autorizaciones para la realización de ensayos clínicos con medicamentos. Esa directiva, poco a poco, se va a ir trasladando a las diversas legislaciones nacionales. Así, por ejemplo, a nivel español, el Real Decreto, 223/20047, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos incorporó en su totalidad al ordenamiento jurídico interno la Directiva 2001/20/CE.
Posteriormente, las legislaciones sobre investigación han asumido como propios los principios de la declaración de Helsinki, es el caso de la Directiva 2005/28/CE sobre buenas prácticas en los procesos de investigación que acoge en su articulado de forma expresa (artículo 3.1) “que se deben respetar la Declaración de Helsinki sobre los principios éticos para las investigaciones médicas en seres humanos”. En España, la Ley 29/2006, de 26 de julio, de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios8, señala también que los ensayos clínicos deberán realizarse siguiendo los contenidos de la Declaración de Helsinki, por lo que esta segunda directiva europea queda incluida en la legislación española.
Actualmente, tanto la Ley de Ensayos Clínicos con Medicamentos, Real Decreto 223/2004 y la Ley 29/2006 de 26 de julio, de Garantías y Uso Racional de los Medicamentos y Productos Sanitarios, son las que definen los aspectos éticos de las investigaciones en seres humanos.
Esta legislación ha sido completada el año 2007 con la Ley de Investigación Biomédica (LIB)9, en la que se establece un control ético obligatorio para todo proyecto de investigación que implique intervenciones en seres humanos o la utilización de muestras biológicas de origen humano. La Ley 14/2007, de 3 de julio, de Investigación Biomédica, establece que “la autorización y desarrollo de cualquier proyecto de investigación sobre seres humanos o su material biológico requerirá el previo y preceptivo informe favorable del Comité de Ética de la Investigación”10 (hay otros aspectos que también regula como la investigación con embriones y preembrioones, los biobancos, etc., pero que omitimos porque exceden del objetivo de este trabajo).
La Ley de Investigación Biomédica encomienda al Comité de Bioética de España (en el artículo 78 apartado c) “establecer los principios generales para la elaboración de códigos de buenas prácticas de investigación científica” (CBPC). Con el fin de cumplir dicho mandato, el Comité de Bioética de España ha publicado unas “Recomendaciones para la implantación de buenas prácticas científicas”11.
En dichas recomendaciones se enumeran los contenidos básicos que debe incluir cualquier CBPC, la implantación de CBPC en el sistema público de ciencia-tecnología-innovación, la formación y fomento de valores en buenas prácticas científicas y la creación de un órgano para resolver problemas de integridad científica. Con este último paso, crea un completo entramado legal cuya función es garantizar la participación libre y con garantías sanitarias de las personas en la investigación.
De todas formas, las inspecciones realizadas por el Grupo de Trabajo de Inspección de Buenas Prácticas Clínicas (BPC) de la Agencia Española de Medicamentos y Productos Sanitarios, sobre la realización de ensayos clínicos, demuestran que existen aspectos de mejora en cuanto a la realización de estos y se necesita reforzar la formación de investigadores, monitores y promotores en BPC. Sería interesante dotar a los centros de investigación de más personal destinado exclusivamente a reforzar la actividad del investigador. De todos modos, también concluyen, que las desviaciones que detectaron en las inspecciones no afectaban en su mayoría de forma negativa ni a los pacientes ni a la calidad de los datos del ensayo clínico12.
Tras los puntos previos de los aspectos históricos y legales, nos acercaremos a la forma en que se deben evaluar los aspectos éticos de un proyecto de investigación.
La evaluación de los proyectos de investigación en seres humanos precisa analizar los aspectos metodológicos, éticos y legales de los mismos, y el seguimiento posterior de los estudios una vez han sido autorizados. Se debe velar y proteger los derechos, seguridad y bienestar de los sujetos que participan en un proyecto de investigación y ofrecer garantía pública al respecto.
Los aspectos que se valoran en la evaluación de un proyecto de investigación son los siguientes:
El Código de Ética y Deontología Médica13, elaborado por la Organización Médica Colegial española, del 10 de septiembre de 1999, dedica su artículo 29 a analizar la experimentación médica sobre la persona, exponiendo los siguientes puntos:
Estos principios concuerdan con los principios éticos recogidos en la Declaración de Helsinki, en la que se afirma que es deber del médico proteger la vida, la salud, la dignidad, la integridad, el derecho a la autodeterminación, la intimidad y la confidencialidad de la información personal de las personas que participen en una investigación (art. 11).
El Real Decreto 223/2004 que regula los ensayos clínicos también afirma que en todos los estudios deberá constar de modo explícito que el trabajo cumple con la Ley Orgánica 15/199914, de 13 de diciembre, de protección de datos de carácter personal y con la Declaración de Helsinki de la Asociación Médica Mundial.
La Ley de Investigación Biomédica 14/2007 también indica que tiene como objetivo regular, con pleno respeto a la dignidad e identidad humanas y a los derechos inherentes a la persona, la investigación biomédica.
El Real Decreto 223/2004 sobre ensayos clínicos exige que se debe garantizar la idoneidad de los estudios y la adecuación de la metodología empleada para el fin propuesto. Así, solo deberían realizarse ensayos clínicos cuando los objetivos impliquen un avance sobre el conocimiento científico sobre el ser humano o sobre su estado de salud y siempre que se minimicen los riesgos en los sujetos participantes en él.
La Ley de Investigación Biomédica indica en su artículo 10 que cualquier investigación de carácter biomédico deberá estar científicamente justificada, cumplir con los criterios de calidad científica generalmente aceptados y realizarse de acuerdo con las obligaciones y estándares profesionales adecuados, bajo la supervisión de un investigador científicamente cualificado, siendo evaluada a su finalización.
Uno de los aspectos abordados por el Real Decreto 223/2004 es la información para los pacientes que debe ser lo más asequible posible para cualquier persona que no tenga una formación específica en aspectos sanitarios. Dicha información debe incluir el objetivo y metodología del estudio, los posibles beneficios para el paciente, los posibles efectos secundarios y el carácter voluntario de la participación, así como, la posibilidad de retirarse en cualquier momento de este sin que por ello se altere la relación médico-paciente. Igualmente, deberá constar qué personas tendrán acceso a los datos del paciente y debe hacer referencia a que se respetará la “Protección de Datos de Carácter Personal” (más información sobre los cambios legislativos relativos a la protección de datos, Ley orgánica 3/2018, de Protección de Datos Personales y garantía de los derechos digitales, al final del capítulo).
La Declaración de Helsinki indica que existen poblaciones que son particularmente vulnerables y que necesitan una protección especial. Estas poblaciones incluyen los que no pueden otorgar o rechazar el consentimiento por sí mismos y los que pueden ser vulnerables a coerción o influencia indebida (art. 9). La investigación en una población vulnerable solo se justifica si la investigación responde a las necesidades de salud de esa población y si existen posibilidades razonables de que la población, sobre la que se efectúa la investigación, puede beneficiarse de sus resultados (art. 17).
Siguiendo con la Declaración de Helsinki15, para la investigación médica en la que se utilicen material o datos humanos, el médico debe pedir un consentimiento (art. 25). Cuando el individuo potencial sea incapaz, el médico debe pedir el consentimiento informado a su representante legal (art. 27). Si el individuo es considerado incompetente, pero es capaz de dar su asentimiento a participar o no en una investigación, el médico debe pedirlo, además del consentimiento del representante legal (art. 28).
El Real Decreto 223/2004, por el que se regulan los ensayos clínicos con medicamentos exige a su vez la máxima protección de las poblaciones más vulnerables en la obtención del consentimiento informado.
Si el sujeto es menor de edad se obtendrá el consentimiento informado de los padres o del representante legal del menor. Cuando el menor tenga 12 o más años, deberá prestar su consentimiento para participar en el ensayo. Igualmente hay obligación de poner en conocimiento del Ministerio Fiscal las autorizaciones de los ensayos clínicos cuya población incluya a menores.
La Ley 41/200216 de autonomía del paciente, dice que, en el caso de menores no incapaces ni incapacitados, emancipados o con 16 años cumplidos, no cabe prestar el consentimiento por representación. Son ellos los que deben dar el consentimiento y no sus padres, aunque a los padres se les informará cuando el riesgo sea grande, según el criterio del facultativo, y su opinión será tenida en cuenta para la toma de la decisión. Pero dicha ley excluye de esta salvedad a los ensayos clínicos, que se rigen los lo establecido con carácter general sobre la mayoría de edad (18 años). Por lo tanto, la doctrina del “menor maduro”, la idea de que a los 16 años existe una “mayoría de edad sanitaria”, en el caso de los ensayos clínicos no tiene lugar17,18.
La ley de Investigación Biomédica 14/2007 en su artículo 4 dice que las personas incapacitadas y los menores participarán en la medida de lo posible y según su edad y capacidades en la toma de decisiones a lo largo del proceso de investigación. En el artículo 20 es más explícito en relación con las personas que no tienen capacidad para expresar su consentimiento (menores o incapaces) y dice lo siguiente:
La investigación sobre una persona menor o incapaz solo podrá realizarse si concurren las siguientes condiciones:
Por lo tanto, en investigaciones que no tuvieran las características de ensayos clínicos sí tendrían lugar las normas relativas al menor maduro, siendo este quien otorgaría su consentimiento al ser de aplicación la ley 41/2002 de autonomía del paciente, que considera, como ya hemos recogido, la mayoría de edad sanitaria, a excepción de ensayos clínicos, a los 16 años.
Entonces habría diferente regulación según el tipo de estudio:
De todas formas, en la hoja de información del paciente que participa en un ensayo clínico debe constar un apartado en el que diga que cuando cumpla 18 años podrá revocar el consentimiento informado otorgado por sus padres.
Las hojas de información al paciente deben realizarse para niños mayores de 12 años, los cuales deben dar su consentimiento. En el caso de estudios de investigación que no sean ensayos clínicos a partir de los 16 años es el propio adolescente el que otorga el consentimiento, sin necesidad de que los padres firmen el documento. Este cambio en el caso de ensayos clínicos se realiza a los 18 años. En menores de 12 años no es preciso el asentimiento del niño, pero si es recomendable realizar una hoja de información adaptada al niño, ya que la legislación indica que “en la medida de su capacidad el niño debe tener información y dar su consentimiento” (Tabla 2).
Tabla 2. Características de la hoja de información al paciente. Mostrar/ocultar
Un aspecto que nos puede tocar desde nuestra actividad en atención pediátrica es que nos consulten aspectos relativos a la investigación en relación con el embarazo y lactancia. De forma similar a lo visto para la investigación en menores, tanto el Real Decreto de Ensayos clínicos como la Ley de Investigación Biomédica abordan estos aspectos.
El Real Decreto 223/2004 establece que en mujeres gestantes o en periodo de lactancia, solo se podrán realizar ensayos clínicos sin beneficio potencial directo para ellas cuando se concluya que no suponen ningún riesgo previsible para su salud ni para la del feto o niño, y que se obtendrán conocimientos útiles y relevantes sobre el embarazo o la lactancia.
Según la Ley de Investigación Biomédica 14/2007, solo podrá realizarse una investigación en una mujer embarazada cuando el objetivo de la investigación sea buscar un beneficio de otras mujeres embarazadas, sobre los fetos o los niños lactantes, cuando no sea posible realizar una investigación similar en mujeres no embarazadas o lactantes y que la mujer embarazada, lactante o los representantes legales del niño, en su caso, presten su consentimiento.
Los límites puestos para la investigación en la infancia son beneficiosos en la medida que protegen a los niños de experimentación poco ética. Aunque se da la paradoja de que, si no se realizan estudios en niños, los medicamentos que utilizamos en ellos no tienen una indicación específica para la infancia.
En una revisión sobre aspectos éticos de la investigación infantil se recoge el dato de que, por las dificultades que lleva la realización de ensayos clínicos en niños, el 70% de los fármacos autorizados por la FDA no incluyen autorización para su uso en niños y en nuestro medio carecen de indicación para niños el 90% de los fármacos utilizados en Cuidados Intensivos Neonatales, el 70% en Cuidados Intensivos Pediátricos, el 50% en Digestivo, el 47% en Cardiología el 40% en Psiquiatría y el 38% en Pediatría General19.
Surge la paradoja de que por tratar de proteger a los niños les estamos privando de una medicación adecuada para ellos. La Dra. Inés Galende en las Jornadas sobre Ensayos Clínicos en Pediatría realizadas en Valencia en 2009 decía que la investigación en los niños debe considerarse como un derecho de estos, con el fin de asegurar una terapia apropiada y basada en datos científicos para la población infantil20. Así, se puede constatar en la Declaración de Helsinki (Seul, 2008), que igual que limita la investigación en poblaciones vulnerables (Art 17: La investigación en poblaciones vulnerables solo se justifica si existen posibilidades razonables de que la población sobre la que se realiza le investigación podrá beneficiarse), propone la participación en ensayos clínicos de las poblaciones infrarrepresentadas (Art. 5: las poblaciones infrarrepresentadas en la investigación médica deben tener un acceso apropiado a la participación en la investigación). Los niños son una de esas poblaciones infrarrepresentadas. Según los datos de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) en el periodo 2004-2005 los ensayos clínicos en niños suponían, tan solo, un 3,2% del total21, lo que es totalmente insuficiente dada la enorme cantidad de medicamentos que se administran fuera de ficha técnica en la edad infantil.
Para tratar de resolver este problema el Parlamento europeo en 2006 publicó un reglamento con la finalidad de facilitar el desarrollo y la accesibilidad de medicamentos de uso pediátrico y propone asimismo la creación de planes de investigación pediátrica cuyo objetivo es demostrar la calidad seguridad y eficacia de los medicamentos en la población pediátrica22. Una revisión realizada por la AEMPS23, tras la aprobación de este reglamento, constata que el número de ensayos clínicos y planes de investigación pediátrica en España ha aumentado en los últimos años. Se ha pasado de 50 ensayos clínicos en el 2007 a 60 en 2008 y a 80 en 2009. El incremento más llamativo ha sido en los ensayos clínicos incluidos en planes de investigación pediátrica que ha pasado de 12 en 2007 a 45 en 2009. De todas formas, la mayoría de los estudios se dan en adolescentes (45% en niños de entre 12 y 17 años) seguido de los niños (35% de entre 2 y 11 años), lactantes (15% en menores de dos años excluyendo el periodo neonatal) y neonatos (5%, incluyendo tanto los niños a término como los pretérmino), por lo que queda el periodo neonatal como aquel en el que menor número de ensayos clínicos se realizan. De todas formas, el porcentaje de ensayos clínicos con población infantil ha aumentado. Si antes veíamos con los datos de la AEMPS de 2005 que el porcentaje de ensayos clínicos en población infantil era del 3,2%, la revisión, realizada en 201024, constata un 11% de ensayos con población infantil, lo que supone un tímido avance.
La documentación que hay que presentar para que un proyecto de investigación sea aprobado depende de las características del proyecto. Los tipos de investigación se dividen en tres tipos principales: ensayos clínicos, proyectos de investigación y estudios de tipo observacional con medicamentos o posautorización (EPA). Veremos cada uno de ellos.
Los ensayos clínicos son los tipos de estudios más exigentes tanto en cuanto a metodología como a exigencias por parte de las evaluaciones éticas. Basándonos en las exigencias del Comité Ético de Euskadi, los documentos necesarios aparecen en la Tabla 3.
Tabla 3. Presentación de ensayos clínicos. Mostrar/ocultar
Los proyectos de investigación tienen menores exigencias. En un proyecto de investigación que no precisara la obtención de muestras biológicas, no sería preciso la existencia de seguro, que como hemos visto es necesario en el caso de los ensayos clínicos, pero según la Ley de Investigación Biomédica, “La realización de una investigación que comporte un procedimiento invasivo en seres humanos exigirá el aseguramiento previo de los daños y perjuicios” (Tabla 4).
Tabla 4. Presentación de proyectos de investigación. Mostrar/ocultar
Si los ensayos clínicos son muy escasos, este tipo de estudios en niños, lo son todavía más. Requieren además de la aceptación del comité ético, el dictamen favorable de la Dirección General de Farmacia. Dichos estudios pueden tener carácter prospectivo o retrospectivo. Los documentos que hay que presentar se resumen en la Tabla 5.
Tabla 5. Presentación de estudios posautorización (EPA). Mostrar/ocultar
Las características legales del consentimiento informado, un pilar de la participación libre de las personas en los estudios de investigación se ha modificado a raíz de la directiva del Parlamento europeo 2016/67925 y la posterior Ley orgánica española 3/2018 de protección de datos personales y garantía de los derechos digitales26.
Con el fin de facilitar a los investigadores biomédicos el conocimiento de esta legislación, la Agencia Española de Protección de Datos27 ha publicado un documento en el que trata de informar a la comunidad científica de estas cuestiones. Estos son las explicaciones principales que expone:
En el modelo vigente, hasta la puesta en marcha de estas nuevas normativas, se establece, como regla general, la necesidad del consentimiento del sujeto para una investigación concreta y se indica que se deberá volver a solicitar un nuevo consentimiento para una futura investigación, salvo determinados supuestos, bien por no ser posible la identificación del sujeto por haber sido anonimizados sus datos, previo dictamen favorable del Comité Ético de Investigación, bien cuando se trate de una investigación relacionada con la inicial, al considerarse el fin de dicha investigación compatible con el de aquella en que se prestó el consentimiento.
Por lo tanto, estaba limitado el consentimiento a la finalidad con la que se prestó. La nueva normativa queda mucho más abierta. Así, señala que con frecuencia no es posible determinar totalmente la finalidad del tratamiento de los datos personales con fines de investigación científica en el momento de su recogida.
Es por ello que la prestación del consentimiento no puede ser interpretada de un modo restrictivo, limitado a una concreta investigación de la que se facilite toda la información disponible, sino que puede extenderse en el futuro ese consentimiento, sin que ello lo vicie en modo alguno, incluso a “finalidades” o áreas de investigación que ni siquiera hubieran podido determinarse en el momento en que se prestó el consentimiento sin que sea necesario recabar uno nuevo, por los beneficios que pueden derivarse para los individuos y la sociedad en su conjunto, que pueden derivarse de la investigación, inicialmente no prevista.
Los aspectos éticos de la investigación biomédica tienen como objetivo fundamental proteger a las personas que participan en una investigación a la vez que garantizar que la decisión de participación sea libre y no tenga repercusiones negativas para su salud.
El niño, que es objeto de nuestra atención clínica, tiene unas limitaciones especiales ya que existe una limitación en cuanto a la capacidad de otorgar su consentimiento que hay que tener en cuenta.
La investigación biomédica de calidad solo puede ser aquella que acredite el respeto a los principios éticos y jurídicos que ha consensuado la sociedad en la que se desarrolla.
Texto actualizado que resume todos los aspectos del control ético de la investigación biomédica.
Gorrotxategi Gorrotxategi P. Aspectos éticos de la investigación biomédica. FAPap Monogr. 2021;6:55-65